





















































































 免費參與·100+跨境活動
免費參與·100+跨境活動 
 免費下載·4000+跨境資料
免費下載·4000+跨境資料 
 免費學習·2000+直播課程
免費學習·2000+直播課程 
 免費加入·15萬+賣家交流群
免費加入·15萬+賣家交流群 


2020-04-15 16:00

紅外體溫計出口通關要求及部分國家(地區)主要技術性貿易措施提示如下:
出口通關要求
紅外體溫計根據其是否與人體表面接觸分為接觸式紅外體溫計(耳溫槍)、非接觸式紅外體溫計(額溫槍)兩種。
商品歸類

禁限管理
根據商務部 海關總署 國家藥品監督管理局2020年第5號《關于有序開展醫療物資出口的公告》:
自4月1日起,出口新型冠狀病毒檢測試劑、醫用口罩、醫用防護服、呼吸機、紅外體溫計的企業向海關報關時,須提供書面或電子聲明,承諾出口產品已取得我國醫療器械產品注冊證書,符合進口國(地區)的質量標準要求。
海關憑藥品監督管理部門批準的醫療器械產品注冊證書驗放。上述醫療物資出口質量監管措施將視疫情發展情況動態調整。

國家藥監局官網醫療器械產品注冊信息查詢地址(動態更新):
http://www.nmpa.gov.cn/WS04/CL2582/
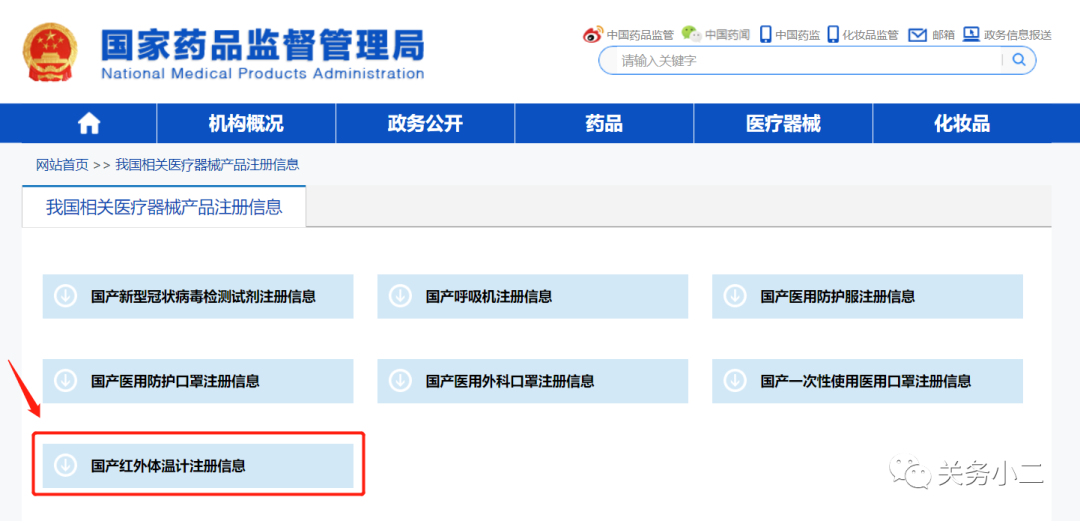
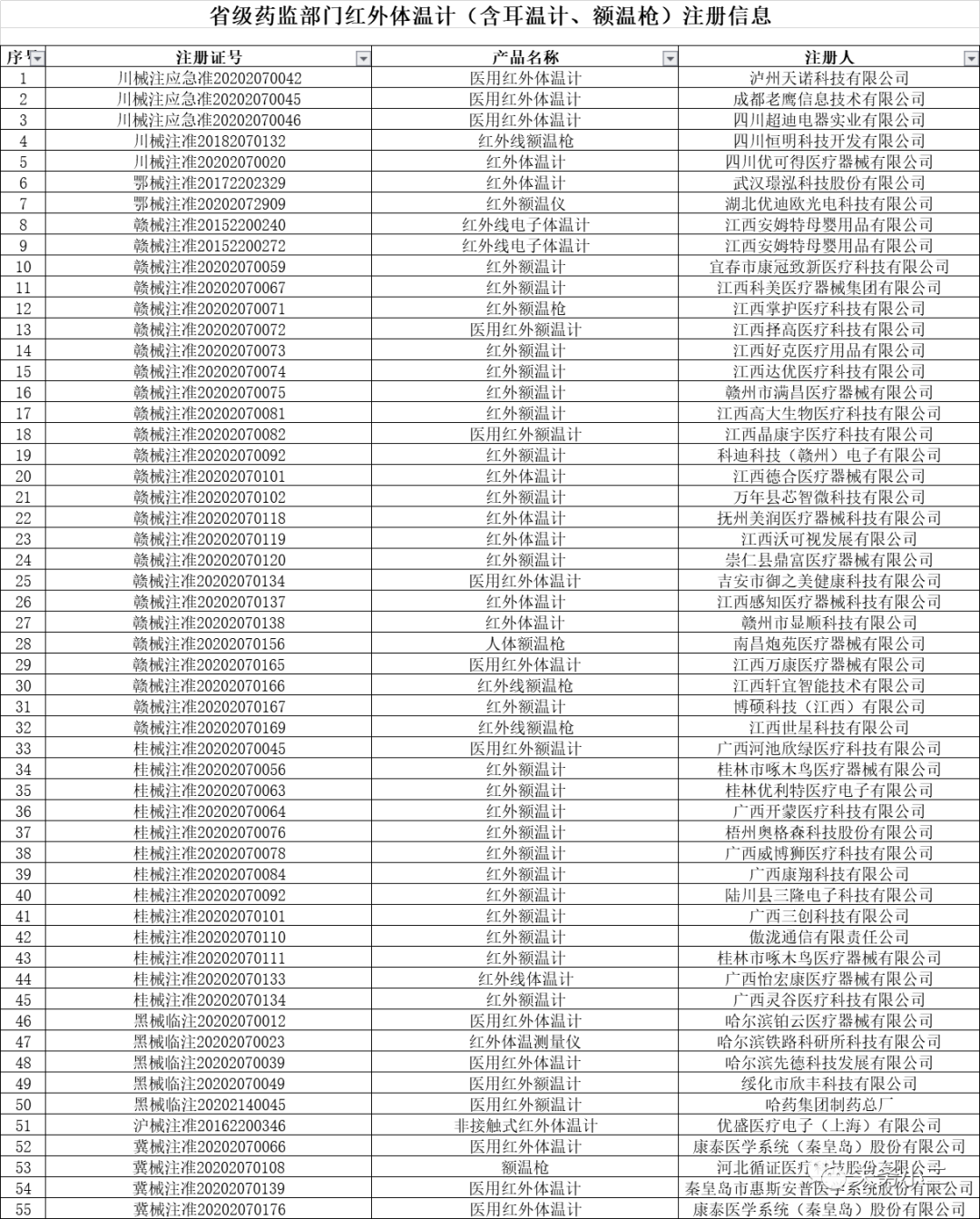
(具有紅外體溫計出口資質的部分企業名單)
退稅管理
紅外體溫計及其配件的出口退稅率為13%。
各國紅外體溫計的準入條件
一美國
醫療器械進入美國市場可按照豁免、510(k)、PMA三種情況申請。常見的紅外體溫計在美國FDA屬于II類醫療器械,需要按照510(k)來申請,510(k)申請流程:
1.進行產品測試(性能測試、生物學測試)
2.準備510(k)技術文件,提交FDA審評
3.獲得FDA的510(k)批準信
4.完成工廠注冊和器械列名
510(k)文件是企業向FDA遞交的產品上市前申請文件,目的是證明所申請上市的產品和已在美國市場上合法銷售的產品在安全性和有效性方面比較是實質相等的,即為等價器械(substantially equivalent)。申請者必須把申請上市的器械與現在美國市場上一種或多種相似器械進行對比,得出支持等價器械的結論。對FDA 510(k)注冊文件所必須包含的信息,FDA有一個基本的要求,其內容大致如下16個方面:
1.申請函。此部分應包括申請人(或聯系人)和企業的基本信息、FDA 510(k)遞交的目的、申請上市器械的名稱型號和分類資料、進行實質等效比較的產品(Predicate Device)名稱及其510(k)號碼。
2.目錄。即FDA 510(k)文件中所含全部資料的清單(包括附件)。
3.真實性保證聲明。FDA有一個標準的樣本。
4.器材名稱。即產品通用名、FDA分類名、產品貿易名。
5.注冊號碼。如企業在遞交FDA 510(k)時已進行企業注冊,則應給出注冊信息,若未注冊,也予注明。
6.分類。即產品的分類組、類別、管理號和產品代碼。
7.性能標準。產品所滿足的強制性標準或自愿性標準。
8.產品標識。包括企業包裝標識、使用說明書、包裝附件、產品標示等。
9.實質相等性比較(SE)。選擇合適的產品進行比較是510(k)申請的關鍵步驟。在進行比較時應從如下方面進行考慮:企業必須提供充足的資料證明,所申請上市的器械和被比較的器械是實質相等的(SE),否則510(k)申請不會通過。
10.510(k)摘要或聲明。申請文件摘要和支持等價器械的結論。
11.產品描述。包括產品的預期用途、工作原理、動力來源、零組件、照片、工藝圖、裝配圖、結構示意圖等。
12.產品的安全性與有效性。包括各種設計、測試資料。
13.生物相容性。生物相容性是指材料與生物體之間相互作用后產生的各種生物、物理、化學等反應的一種概念。一般地講,就是材料植入人體后與人體相容程度,也就是說是否會對人體組織造成毒害作用。
14.色素添加劑(如適用)。
15.軟件驗證(如適用)。
16.滅菌(如適用)。包括滅菌方法的描述、滅菌驗證產品包裝和標識等。
美國的紅外體溫計相關檢測標準包括:
ANSI/AAMI ES60601-1:2005/(R)2012,(IEC 60601-1:2005,MOD)《醫用電氣設備第1部分:基本安全和基本性能的通用要求》,ANSI/AAMI/IEC 60601-1-2:2014《醫用電氣設備第1-2部分:安全通用要求并列標準:電磁兼容要求和試驗》,ANSI/AAMI HA 60601-1-11:2015 (IEC 60601-1-11:2015,MOD)《醫用電氣設備第1-11部分:基本安全和基本性能的通用要求.并列標準:醫用電氣設備和家庭醫療保健環境中使用的醫用電氣系統的要求》,ASTM E 1965-98:2016《間歇測定病人體溫用的紅外溫度計》。按檢測類型分為基本要求、電磁兼容要求和性能要求。
技術標準簡析:

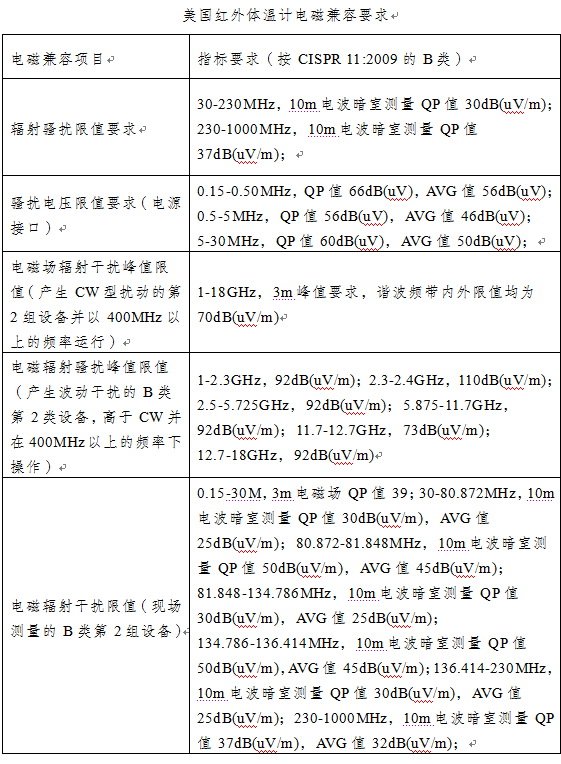

美國紅外體溫計涉及的項目包括輸入功率;保護接地、功能接地和電位均衡;電介質強度;抗跌落性;電磁兼容等。項目要求如下:
二歐盟
歐盟將醫療器械劃分為三大類,I類(包含I和I*)、II類(包含IIa和IIb)及III類。無論屬于哪一類,出口歐盟都需要申請CE認證。
紅外體溫計屬于II類醫療器械,II類以上產品需要歐盟公告機構參與,如TUV南德、TUV萊茵、BSI、DNV、SGS等。企業需要先完成CE認證文件并提交通過審核后方進行體系審核,最終拿到CE和符合歐盟標準的體系證書。
紅外體溫計的CE認證需要滿足歐盟醫療器械指令93/42/EEC(MDD)或歐盟醫療器械條例EU2017/745(MDR)。
紅外體溫計的CE認證程序如下:
1.企業向認證機構提出認證申請并填寫認證詢價單交認證機構。
2.認證機構向申請認證企業提出報價單,企業簽字確認即完成合約。
3.企業向認證機構提交ISO9000+ISO13485質量體系文件即質量手冊和程序文件,供認證機構進行體系文件審核;質量體系審核前,企業應至少三個月的質量體系運行記錄,并完成1-2次內部質量體系審核。
4.認證機構發出認證產品測試通知單給認證機構認可的實驗室,由實驗室對申請認證的產品進行測試。測試合格后,實驗室出具試驗報告。相關的檢測標準如下:
EN 60601-1:2006+A1:2013《醫用電氣設備第1部分:基本安全和基本性能的通用要求》
EN 60601-1-2:2015《醫用電氣設備 第1-2部分:安全通用要求 并列標準:電磁兼容 要求和試驗》
EN 60601-1-11:2015《醫療電氣設備 第1-11部分:基本安全和基本性能的一般要求 并行標準:家用醫療保健環境使用的醫療電氣設備和醫療電氣系統的要求》
ISO80601-2-56:2017《醫用電氣設備 第2-56 部分:體溫測量的臨床體溫計基本安全和基本性能的特殊要求》
5.制造商必須根據產品所符合指令的要求及風險評估的需要,建立產品的技術文件(TCF)。如果相關授權部門要求,制造商須將技術文件及EC符合性聲明一起提交檢查。
6.認證機構對企業的ISO9000+ISO13485質量體系和TCF文件進行審核。
7.正式審核通過后,認證機構將于企業簽訂框架協議,明確各方應遵循原則和CE標志的范圍等。
建議企業找歐盟認可的公告機構進行檢測:
1.歐盟官網MDD 93/42/EEC醫療器械指令授權的機構查詢地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=13
2.歐盟官網MDR (EU) 2017/745醫療器械法規授權的機構查詢地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=34
技術標準簡析:
歐盟和美國標準基本要求都采標自國際標準IEC 60601-1:2005(歐盟標準采標自IEC 60601-1:2005+A1:2012),IEC 60601-1-11:2015;電磁兼容要求都采標自國際標準IEC 60601-1-2:2014。故基本要求和電磁兼容要求無大差異,詳見之前美國技術標準解析,但歐盟性能標準與美國有差異。
歐盟紅外體溫計性能要求:

三日本
如是需要投放市場產品,必須滿足日本的Pharmaceutical and Medical Device Act (PMD Act),在PMD Act的要求下,TOROKU注冊系統要求國外的制造商必須向日本醫藥品和醫療器械綜合機構(PMDA)注冊制造商信息。
日本醫藥品和醫療器械綜合機構網址:www.pmda.go.jp(需復制并通過瀏覽器打開)。
四韓國
韓國衛生福利部(Ministry of Health and Welfare),簡稱衛生部MHW,主要負責管食品、藥品、化妝品和醫療器械的管理,是最主要的衛生保健部門。依照《醫療器械法》,韓國衛生福利部下屬的食品藥品安全部負責對醫療器械的監管工作。KFDA注冊流程為:
1.確定產品分類(I,II,III,IV),選擇KLH(韓國持證方)。
2.II類產品需申請KGMP證書和接受現場審核,II類產品一般是授權的第三方審核員,并獲得KGMP證書。
3. II類產品需要送樣品到韓國MFDS授權的實驗室進行韓國標準的測試。
4.由KLH向MFDS(韓國食品藥品安全部)提交技術文件(檢測報告,KGMP證書等),進行注冊審批。
5.支付申請費用。
6.注冊文件整改,注冊批準。
7.指定韓國代理商和經銷商,產品銷售。涉及標準有:
KS C ISO 80601-2-56:2012《醫用電氣設備 第2-56 部分:體溫測量的臨床體溫計基本安全和基本性能的特殊要求》
KS C IEC 60601-1:2011 《醫用電氣設備第1部分:基本安全和基本性能的通用要求》
KS C IEC 60601-1-2:2012《醫用電氣設備 第1-2部分:安全通用要求 并列標準:電磁兼容 要求和試驗》
KS C IEC 60601-1-11:2012《醫療電氣設備.第1-11部分:基本安全和基本性能的一般要求.并行標準:家用醫療保健環境使用的醫療電氣設備和醫療電氣系統的要求》
KS ISO 10993-5:1999《醫療器械生物學評估-第5部分:體外細胞毒性試驗》
KS ISO10993-10:2002《醫療器械生物學評估-第10部分:刺激性和延遲過敏反應試驗》
韓國藥監局Korea Pharmaceutical Traders Association網址:www.kpta.or.kr(需復制并通過瀏覽器打開)。
五澳大利亞
在澳大利亞,醫療器械是指用于人體的儀器、設備、器具、材料或者其他物品(單獨或者組合使用及適當應用所需的軟件),用以實現診斷、預防、監護、治療或緩解疾病的目的。
在澳大利亞生產醫療器械產品,必須通過合格性評定,評定醫療器械是否符合在相關國家市場(如澳大利亞、歐洲及美國)上市的標準要求;銷售醫療器械則必須在器械電子申請報關系統(DEAL)進行在線申請,完成在澳大利亞治療品注冊處(ARTG)的產品注冊。ARTG是注冊處的一個治療產品備案制度,要求經銷商對所生產和經營(包括進出口)的醫療器械產品負責。
在澳大利亞生產經營醫療器械產品,本土生產商需獲得治療品管理局頒發的合格評定證書;海外生產商則需要具備互認協議證書/歐洲CE認證和澳大利亞的合格聲明。生產商將合適的合格評定證書交給經銷商,再由經銷商通過DEAL系統進行提交作為生產商資質的證明。TGA審評后將通過生產商的資質證明申請。經銷商通過DEAL系統提交醫療器械申請并繳納一定申請費用,TGA會把該醫療器械添加到ARTG備案中。經銷商必須繳納年費,TGA需對產品進行上市后的監測。
海外生產商需具備互認協議證書/歐洲CE認證和澳大利亞的合格聲明。
各國紅外體溫計技術標準簡析
??各國家(地區)關于紅外體溫計等醫療器械相關的技術標準為各國家(地區)進口或銷售時,由其海關或相關部門要求驗核;如有動態調整,請以相關標準管理機構官方發布為準。
??您也可登陸海關總署商品檢驗司網站“政策法規”欄目查詢最新標準(復制并通過瀏覽器打開:http://sjs.customs.gov.cn/)。
(來源:關務小二)
以上內容屬作者個人觀點,不代表雨果網立場!如有侵權,請聯系我們。
















