





















































































 免費參與·100+跨境活動
免費參與·100+跨境活動 
 免費下載·4000+跨境資料
免費下載·4000+跨境資料 
 免費學習·2000+直播課程
免費學習·2000+直播課程 
 免費加入·15萬+賣家交流群
免費加入·15萬+賣家交流群 


2020-04-02 10:46

技術貿易措施
為統籌做好新型冠狀病毒肺炎疫情防控和經濟社會發展工作重要指示,落實海關總署支持外貿企業復工復產,促外貿穩增長10項措施,結合國內外新冠肺炎疫情防控形勢、國內防護物資產能過剩、外貿企業頻繁咨詢防護物資出口政策等因素,為促進外貿企業復工復產、有效消化國內富余產能和富余物資,促外貿穩增長,現就物資出口通關要求及收集整理的呼吸機等防控物資國外主要技術性貿易措施提示如下:
在現代臨床醫學中,呼吸機作為一項能人工替代自主通氣功能的有效手段,已普遍用于各種原因所致的呼吸衰竭、大手術期間的麻醉呼吸管理、呼吸支持治療和急救復蘇中,在現代醫學領域內占有十分重要的位置。呼吸機是一種能夠起到預防和治療呼吸衰竭,減少并發癥,挽救及延長病人生命的至關重要的醫療設備。
01
出口通關要求
商品歸類
商品
說明
商品編號
呼吸機
臭氧治療器、氧氣治療器、噴霧治療器、人工呼吸器或其他治療用呼吸器具
9019.2000
禁限管理
根據商務部 海關總署 國家藥品監督管理局2020年第5號《關于有序開展醫療物資出口的公告》。鏈接:http://www.mofcom.gov.cn/article/b/e/202003/20200302950371.shtml
為有效支持全球抗擊疫情,保證產品質量安全、規范出口秩序,自4月1日起,出口新型冠狀病毒檢測試劑、醫用口罩、醫用防護服、呼吸機、紅外體溫計的企業向海關報關時,須提供書面或電子聲明,承諾出口產品已取得我國醫療器械產品注冊證書,符合進口國(地區)的質量標準要求。海關憑藥品監督管理部門批準的醫療器械產品注冊證書驗放。上述醫療物資出口質量監管措施將視疫情發展情況動態調整。
有關醫療物資出口企業要確保產品質量安全、符合相關標準要求,積極支持國際社會共同抗擊疫情。

目前公告的具有呼吸機出口資質的企業名單如下:
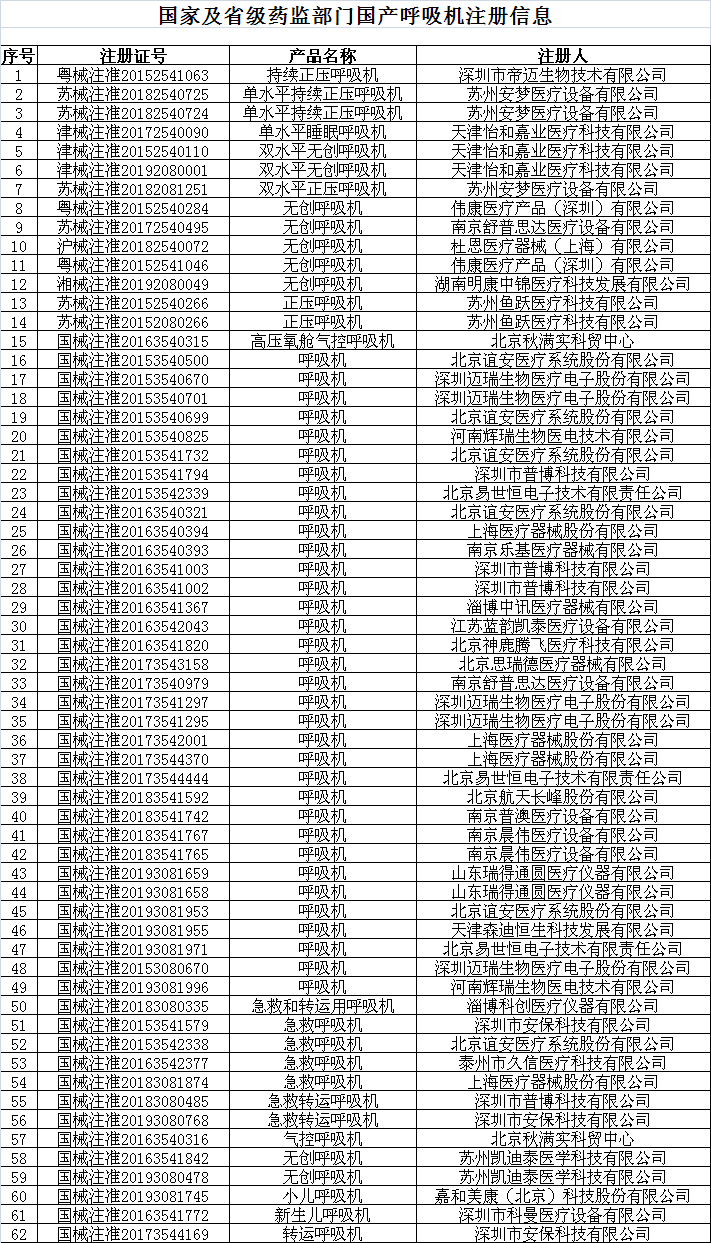
*該名單在國家藥監局網站www.nmpa.gov.cn動態更新。
退稅管理
呼吸機及其配件的出口退稅率為13%。
↓↓↓以下內容是根據國內外相關政府機構、↓↓↓
專業網站、新聞報道收集整理而成,
僅供參考。具體內容以相關管理部門、
國外官方機構要求為準。
相關貨物出口時,中國海關無下列相關證件要求。
02
各國呼吸機準入條件
美國
必須要取得美國食品和藥物管理局FDA注冊認證才可以在美國本土市場進行銷售,且FDA注冊認證必須在美國本土辦理,建議企業申請時委托美國本地代理。
美國宣布自3月11日起取消部分醫療物資進口關稅,其中包括呼吸機。企業可通過網址https://hts.usitc.gov/進行驗證相關產品稅率。
美國FDA對醫療器械的管理是通過器械與放射健康中心(CDRH)進行的,其對每一種醫療器械的分類和管理要求極其嚴格。根據風險等級的不同,FDA將醫療器械分為三類(Ⅰ,Ⅱ,Ⅲ),呼吸機屬于Ⅲ類高風險等級醫療設備。在美國上市的呼吸機,須按照企業注冊、產品列名、一般控制、特殊控制,向FDA遞交510(k)等步驟逐步提交全部技術和臨床文件,以期認證呼吸機的安全性、可靠性以及各項參數、性能等諸多方面,最終通過嚴謹的論證和試驗來證明申請認證的機型可以完全滿足FDA 的要求。
FDA 510(k)為醫療器械在美國上市的主要途徑之一,絕大多數的II類醫療器械和部分I類、III類醫療器械通過此途徑清關上市。510(k)文件是向FDA遞交的上市前申請文件,目的是證明所申請上市的產品和已在美國市場上合法銷售的產品在安全性和有效性方面比較是實質相等的,即為等價器械(substantially equivalent)。申請者必須把申請上市的器械與現在美國市場上一種或多種相似器械對比,得出并且支持等價器械的結論。對FDA 510(k)注冊文件所必須包含的信息,FDA有一個基本的要求,其內容大致如下16個方面:
1.申請函
此部分應包括申請人(或聯系人)和企業的基本信息、FDA 510(K)遞交的目的、申請上市器械的名稱型號和分類資料、進行實質等效比較的產品(Predicate Device)名稱及其510(k)號碼。
2.目錄
即FDA 510(k)文件中所含全部資料的清單(包括附件)。
3.真實性保證聲明
FDA有一個標準的樣本
4.器材名稱
即產品通用名、FDA分類名、產品貿易名。
5.注冊號碼
如企業在遞交FDA 510(k)時已進行企業注冊,則應給出注冊信息,若未注冊,也予注明。
6.分類
即產品的分類組、類別、管理號和產品代碼。
7.性能標準
產品所滿足的強制性標準或自愿性標準。
8.產品標識
包括企業包裝標識、使用說明書、包裝附件、產品標示等。
9.實質相等性比較(SE)
選擇合適的產品進行比較是510(K)申請的關鍵步驟。在進行比較時應從如下方面進行考慮:企業必須提供充足的資料證明,所申請上市的器械和被比較的器械是實質相等的(SE),否則510(k)申請不會通過。
10.510(k)摘要或聲明
申請文件摘要和支持等價器械的結論。
11.產品描述
包括產品的預期用途、工作原理、動力來源、零組件、照片、工藝圖、裝配圖、結構示意圖等。
12.產品的安全性與有效性
包括各種設計、測試資料。
13.生物相容性
生物相容性是指材料與生物體之間相互作用后產生的各種生物、物理、化學等反應的一種概念。一般地講,就是材料植入人體后與人體相容程度,也就是說是否會對人體組織造成毒害作用。
14.色素添加劑(如適用)
15.軟件驗證(如適用)
16.滅菌(如適用)
包括滅菌方法的描述、滅菌驗證產品包裝和標識等。
● 技術標準簡析
需特別注意的是,美標中要求嬰兒呼吸機工作壓力控制應在整個范圍內精確至 ±2cm H2O,而其他呼吸機應精確至±5cm H2O。與美標相比,國標沒有按照嬰兒呼吸機和其他呼吸機進行要求,要求讀數的精度為± (2%滿刻度+4%實際讀數)。另外就是國標對報警聲音的要求是最新執行的標準,而美國的設備是按照聲稱的標準進行測試。
歐盟
須獲得歐盟CE認證,并符合技術法規:MDD 93/42/EEC或MDR (EU) 2017/745。
建議找歐盟認可的公告機構進行檢測。
1.歐盟官網MDD 93/42/EEC醫療器械指令授權的機構查詢地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=13

2.自2020年5月26日起,MDR (EU) 2017/745醫療器械法規將正式取代歐盟現行的MDD醫療器械指令強制實施,同樣在歐盟官網可以查詢到。
歐盟官網MDR (EU) 2017/745醫療器械法規授權的機構查詢地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=34

特別提醒:2020年5月26號施行的歐盟MDR對目前CE認證MDD而言有哪些影響?
● 目前大部分CE證書是按照MDD要求測試的,面臨1月后MDR的換證問題;
● 擁有MDD授權的公告機構,并不全是MDR授權的公告機構,CE認證審核機構可選性降低;
● MDR的審核要求比MDD更為復雜,認證周期必然大幅度拉長;
● CE認證費用可能將有大幅提升;
● 歐盟對醫療設備的監管更加嚴格。
日本
如果需要投放市場產品必須滿足日本的Pharmaceutical and Medical Device Act (PMD Act),在PMD Act的要求下,TOROKU注冊系統要求國外的制造商必須向PMDA注冊制造商信息。
日本醫藥品和醫療器械綜合機構(PMDA)網址:www.pmda.go.jp

韓國
韓國醫療器械準入的法規門檻,基本分類為I、II、III、IV類,持證為韓國公司(License holder),韓國收貨人需要到韓國藥監局Korea Pharmaceutical Traders Association. 必須提前備案進口資質。
韓國藥監局Korea Pharmaceutical Traders Association網址:www.kpta.or.kr。

澳大利亞
須通過澳洲的TGA注冊,TGA 是Therapeutic Goods Administration的簡寫,全稱是治療商品管理局。澳大利亞對醫療器械分為I類,Is and Im, IIa, IIb, III類,分別為豁免、備案和注冊。無論哪類醫療器械,其上市銷售前必須得到澳大利亞政府的準許,符合醫療器械的基本要求,按照符合性審查程序進行審查。
特別提醒:澳大利亞已與歐盟達成互認協議。這意味著,合格評定證書由TGA頒發的也被歐盟認可,TGA也認可歐盟CE認證。已獲CE認證的用戶,可提交CE證書及相關資料,獲得TGA證書。
如果產品已經注冊或備案,制造商更換經銷商對其沒有影響。對國外產品進行注冊審批后,每年還要常規注冊一次,說明產品型號、性能及質量有無變化。TGA 全權負責對醫療器械的符合性評價,并收取一定費用,相關費用金額可參見 TGA的網站。
TGA網址:https://www.tga.gov.au/

03
各國呼吸機技術標準簡析
國家(地區)
標準號
標準名稱
美國
ANSI/ASTM/IEC 60601.2.12-2009
醫用電氣設備 第2-12部分:肺呼吸機的特殊安全性要求.ASTM國際標準容許偏差的重癥監護呼吸機
ANSI/ASTM/ISO 10651-4-2002
醫用肺呼吸機 第4部分:操作員控制用人工呼吸機的特殊要求
ANSI/ASTM/ISO 10651-5-2006
醫用肺呼吸機 基本安全和基本性能的特殊要求.第5部分:氣動急救人工呼吸器
ANSI/NFPA 1981-2006
救火和急救服務用開路自持呼吸機標準
ASTM ANS/IEC 60601-2-12-2001
醫用電氣設備 第2-12部分:呼吸機安全特定要求-急救用呼吸機-經ASTM國際批準作為帶差異的美國國家標準
ANSI/ASTM/ISO 10651-4-2ASTM ANS/IEC 60601.2.12-2001
醫療電氣設備 第2-12部分; 肺通氣器安全特別要求—關鍵護理呼吸機通過美國國家標準認證,偏差符合ASTM國際標準
ASTM ANSI/ISO 10651-4-2002
呼吸機
ASTM ANSI/ISO 10651-5-2006
醫用呼吸機 基本安全和基本性能的特定要求
歐盟
EN 794-3-2009
肺呼吸機 第3部分:緊急呼吸機和運輸呼吸機的特殊要求
EN ISO 80601-2-12-2011
醫用電氣設備 第2-12部分:危重護理呼吸機的基本安全和基本性能專用要求
EN ISO 80601-2-72-2015
醫療電氣設備 第2-72部分:對呼吸機依賴患者的家庭醫療保健環境通風機基本安全和基本性能的特殊要求
EN 14048-2002
包裝 測定在氣化介質中包裝材料的最終氧化生物降解性 封閉式呼吸機氧量測量方法
EN 14529-2005
呼吸保護裝置 逃生用包含由需求閥控制的正壓呼吸器的帶半罩式面具的自持式開路壓縮空氣呼吸機
EN 60601-2-12-2006
醫用電氣設備 肺呼吸機安全性特定要求.第2-12部分:危急護理呼吸機
日本
JIS T 7204:1989
醫療用人工呼吸機
韓國
KS C IEC 60601-2-12-2011
醫用電氣設備 肺通氣機安全性的特定要求.第2-12部分:危急護理通氣機
澳大利亞
EN 794-3-2009
肺呼吸機 第3部分:緊急呼吸機和運輸呼吸機的特殊要求
EN ISO 80601-2-12-2011
醫用電氣設備 第2-12部分:危重護理呼吸機的基本安全和基本性能專用要求
EN ISO 80601-2-72-2015
醫療電氣設備 第2-72部分:對呼吸機依賴患者的家庭醫療保健環境通風機基本安全和基本性能的特殊要求
EN 14048-2002
包裝 測定在氣化介質中包裝材料的最終氧化生物降解性 封閉式呼吸機氧量測量方法
EN 14529-2005
呼吸保護裝置 逃生用包含由需求閥控制的正壓呼吸器的帶半罩式面具的自持式開路壓縮空氣呼吸機
EN 60601-2-12-2006
醫用電氣設備 肺呼吸機安全性特定要求.第2-12部分:危急護理呼吸機
俄羅斯
GOST R ISO 80601-2-12-2013
醫療電氣設備. 第2-12部分. 重癥監護呼吸機基本安全和基本性能的特殊要求
國際標準
ISO 10651-6-2004
醫用肺通氣機 基本安全和主要性能的特殊要求 第6部分:家用呼吸機輔助設備
ISO 80601-2-12-2011
醫療電氣設備.第2-12部分:急救護理呼吸機的基本安全性和本質性能的詳細要求
ISO 80601-2-72-2015
醫療電氣設備. 第2-72部分: 對呼吸機依賴患者的家庭醫療保健環境通風機基本安全和基本性能的特殊要求
*以上技術標準為各國進口或銷售時,由各國海關或相關部門要求驗核;如有動態調整,以相關標準管理機構官方發布為準,僅僅作為提醒或參考資料。
*也可至海關總署商品檢驗司子網站查詢,網址:http://sjs.customs.gov.cn/。
供稿單位:廈門海關 上海海關
(來源:12360海關熱線)
以上內容屬作者個人觀點,不代表雨果網立場!如有侵權,請聯系我們。


























